Step 2: Lipoprotein Entry into the Intima
When the Gate Opens

This is the second in a 13-part series exploring the biology of atherosclerosis — from the earliest microscopic changes in the artery wall to the moment a heart attack strikes. Each step connects cutting-edge science to real-world prevention.
In Step 1, we saw how the endothelium, the delicate inner lining of the arteries, can falter and lose its protective grip. Now, in Step 2, we follow what happens next — when ApoB-containing lipoproteins slip through the weakened gate and begin the silent process that will eventually build a plaque.
The Trickle That Starts It All
It never begins with a flood. Atherosclerosis starts with a trickle.
One particle here, another there — silent, unnoticed. They drift along the bloodstream until they find a weak spot in the endothelium. And once the lining falters, the gate opens.
What crosses that threshold are not blobs of dietary fat or grease from last night’s dinner — as old headlines once suggested — but tiny, highly organized couriers: cholesterol-rich lipoproteins carrying a single molecular signature called ApoB.
The image shows an LDL particle and an Lp(a) particle. Both carry cholesterol, but that’s not what makes them atherogenic. What they have in common — and what defines their ability to enter the arterial wall and trigger atherosclerosis — is the presence of a single molecule of ApoB.

The Real Villains
These particles go by many names:
Low-density lipoprotein (LDL): the familiar “bad cholesterol.”
VLDL remnants: leftovers of triglyceride transport, stripped of fat and now smaller, denser, more dangerous.
Intermediate-density lipoprotein (IDL): halfway between VLDL and LDL, but no less of a threat.
Lipoprotein(a) [Lp(a)]: a genetic wildcard, carrying an extra protein tail that makes it unusually sticky.
Different names, different origins — but one common thread: each carries exactly one ApoB molecule. Like a universal passport, ApoB is what allows them to cross the endothelium and take up residence in the arterial wall.
ApoB: Passport and Hook
ApoB (apolipoprotein B) is the structural protein found on every atherogenic lipoprotein. Each particle has one ApoB — no more, no less. That makes ApoB a direct particle count: the higher the ApoB, the more lipoproteins in circulation that can slip into the artery wall.
LDL cholesterol (LDL-C) tells you how much cholesterol is being carried. ApoB tells you how many trucks are on the road. And here, it’s not the size of the cargo that matters — it’s the number of trucks trying to force their way through.
But ApoB isn’t just a marker. It’s the key that allows particles to stay. Once a particle crosses the endothelium, it enters the intima — a layer rich in proteoglycans, long, negatively charged molecules that normally provide structural support and regulate signaling in the vessel wall.
Those proteoglycans — especially biglycan and decorin — are sticky. Their sugar side chains act like hooks, and ApoB carries positively charged domains that snap into place. Free in the bloodstream, LDL is harmless. Once bound in the intima, it is trapped.
And being trapped changes everything. Enzymes and reactive oxygen species remodel the particle. Cholesterol esters are hydrolyzed, phospholipids oxidized, ApoB itself chemically altered. What was once a neutral courier becomes a danger signal, flagged for attack by the immune system.
By contrast, HDL particles don’t carry ApoB. Their surface is built around apolipoprotein A-I, which doesn’t bind proteoglycans. HDL can move in and out of the artery wall freely, sometimes even helping to clear cholesterol — the molecular janitor beside ApoB’s unwelcome intruder.
Size plays a role too. ApoB particles (20–70 nanometers) are small enough to squeeze through endothelial gaps, while much larger chylomicrons from meals are simply too bulky. That’s why last night’s dessert doesn’t directly stick to your arteries. It’s the constant trickle of smaller ApoB particles, day after day, that seeds disease.
ApoB-containing particles bombard the endothelium, with some slipping through and binding to proteoglycans in the intima. In contrast, HDL particles carrying ApoA-I move freely in and out without getting trapped.

A Clinical Puzzle
All of this biology shows up in the clinic in subtle but important ways.
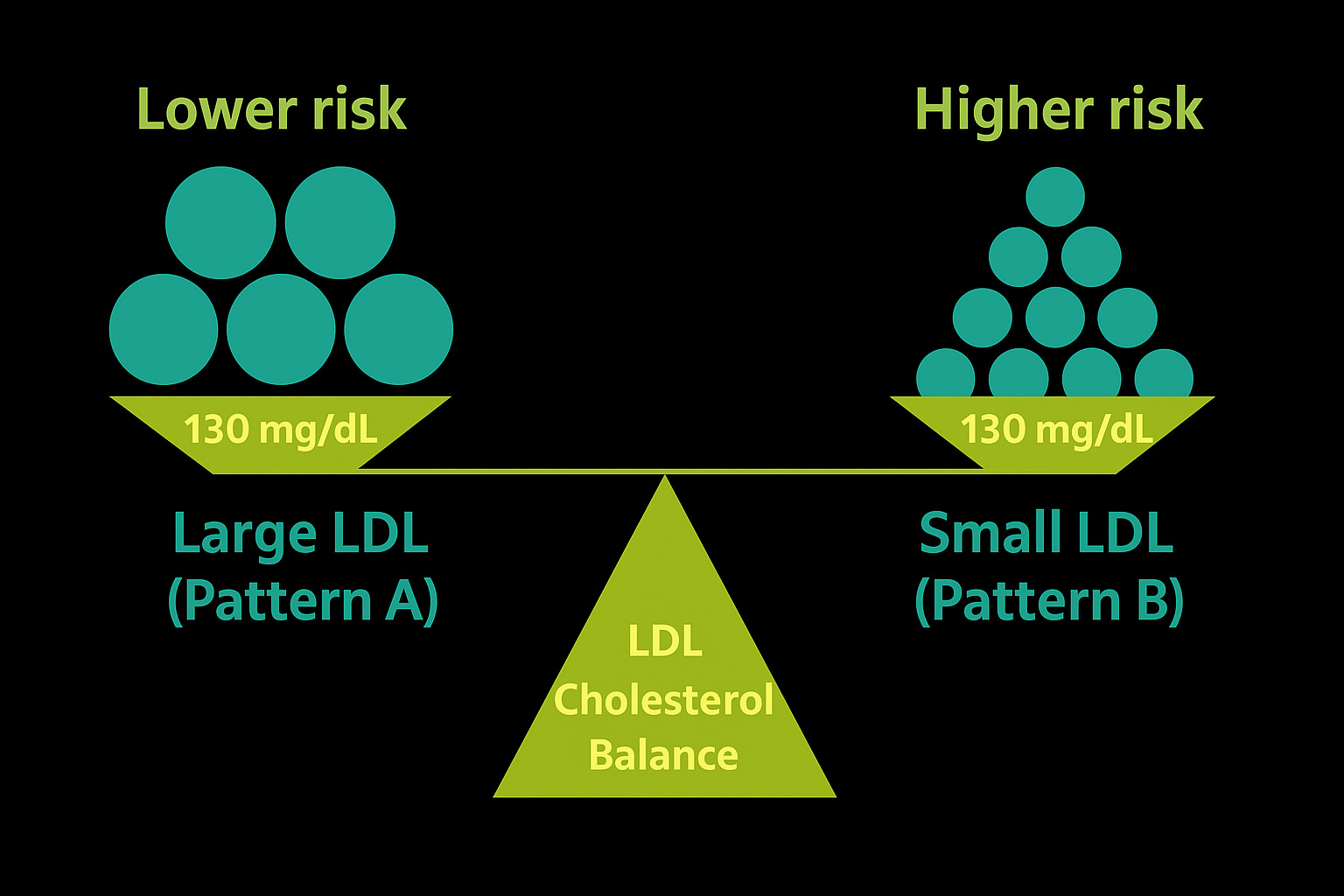
Two patients sit across from you. Both have an LDL-C of 130 mg/dL.
Patient A: ApoB 70 mg/dL.
Patient B: ApoB 110 mg/dL.
On paper, they look identical if you only check LDL-C. But Patient B is carrying thousands more particles — more traffic, more opportunities for invasion. Despite the same cholesterol concentration, their risks are very different.
This is the paradox: cholesterol concentration is not the same as particle number. ApoB gives the count directly and consistently outperforms LDL-C as a predictor of risk.
Two people can have the same LDL-C (130 mg/dL), but very different risks. Large LDL particles (Pattern A) mean fewer particles, while small LDL particles (Pattern B) mean many more. What really reflects this difference is ApoB — a direct count of particle number, and a far better predictor of risk than LDL-C alone.
A Brief Detour Through History
Why, then, has medicine focused for so long on LDL-C and not ApoB?
The answer is partly history, partly convenience.
In the 1970s, laboratory technology made it simple to measure cholesterol concentration. LDL-C was easy, cheap, reproducible, and perfect for large population studies. It became the default currency of cardiovascular risk.
ApoB, by contrast, required specialized assays. It wasn’t until the 1990s and early 2000s that large studies proved its clear superiority. By then, LDL-C was written into textbooks, guidelines, and medical culture. Doctors learned “bad cholesterol.” Patients learned to fear it. Clinical trials were built around it. ApoB — though more biologically meaningful — was sidelined.
That inertia persists. Many labs still don’t report ApoB. Insurers in the U.S. still call it “experimental,” despite decades of evidence.
But science is catching up. In 2021, the European Atherosclerosis Society declared ApoB the “best single marker” of atherogenic lipoproteins. Canadian and European guidelines now reflect that view. The U.S. is still lagging — but the direction of travel is clear.
The Bottom Line
Atherosclerosis does not start in the clinic when a lab report lands on a desk. It starts years earlier, silently, when ApoB-containing particles slip through a weakened endothelium and become trapped in the arterial wall.
LDL-C tells us about the cargo.
ApoB tells us about the invaders.
And it is the invaders that matter.
Each one that stays behind is not just a number on a blood test — it is a seed. A seed that will be oxidized, recognized as foreign, and targeted by the immune system. A seed that will summon inflammation, foam cells, and scar tissue. A seed that, given time, can grow into the very plaque that narrows arteries and ruptures hearts.
This is where the story truly begins. Not in the bloodstream, but in the vessel wall. Not with cholesterol levels, but with the molecular hooks that trap particles where they don’t belong.
👉 Next Step: Retention by Proteoglycans – Stuck Where It Shouldn’t Be