Step 5: Activation of Innate Immunity
Sounding the Alarm

The story of atherosclerosis has been unfolding in stages.
First, the endothelium faltered, leaving a breach in the vessel’s defenses.
Next, lipoproteins slipped inside, sneaking into the intima where they didn’t belong.
Then they became trapped, bound tightly to the sticky matrix of the arterial wall.
Over time, those particles changed — oxidized, modified, and transformed into signals of danger.
Up until now, the immune system has been quiet, watching. But the silence doesn’t last. The body recognizes that something is wrong, and the innate immune system is the first to respond.
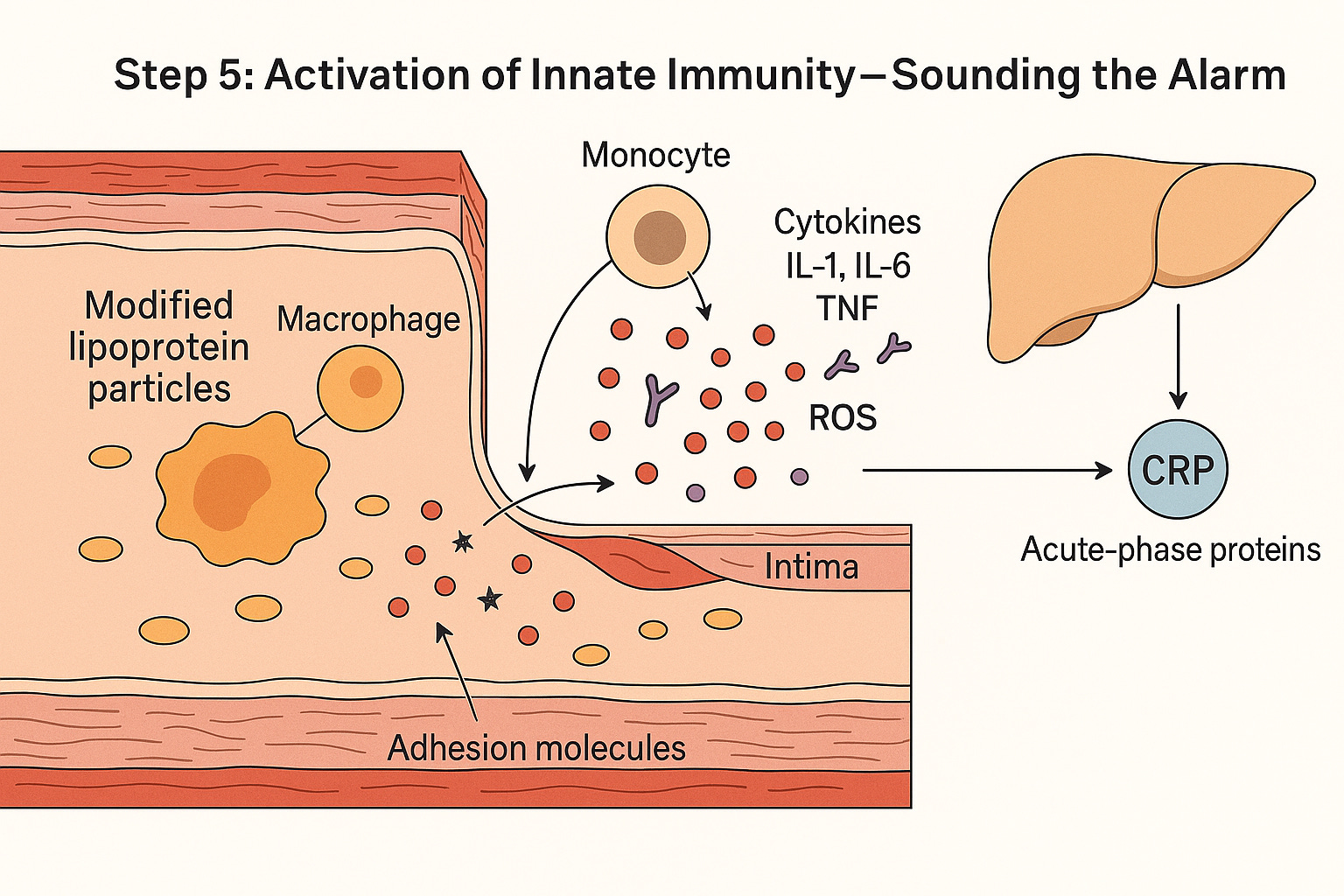
Activation of Innate Immunity — Modified lipoproteins trigger macrophages to release cytokines and reactive oxygen species, recruiting more immune cells and prompting the liver to produce CRP and other acute-phase proteins. A quiet lipoprotein deposit has now become an inflammatory fire.”
Innate Immunity: Always on Duty
Innate immunity is the body’s rapid-response force. Unlike the adaptive immune system, which learns, remembers, and fine-tunes responses, innate immunity doesn’t wait or discriminate. It doesn’t ask who the enemy is — only is there danger?
It works through two main arms: The cellular arm and the humoral arm. Together, these two arms create a system that may not be subtle, but is remarkably effective at sounding the alarm.
The Cellular Arm
The cellular arm of innate immunity is made up of monocytes (which can develop into macrophages in tissues), macrophages, and neutrophils. These are the ‘first responder’ cells, always patrolling the body and ready to detect trouble.”
What makes them effective are their pattern-recognition receptors (PRRs) — molecular sensors that act like security scanners.
Toll-like receptors (TLRs): Sit on the cell surface or in little pockets inside the cell (endosomes). They pick up signals from microbes or damaged molecules.
NOD-like receptors (NLRs): Reside in the cytoplasm, monitoring for danger inside the cell. They can switch on inflammasomes — powerful alarm systems.
RIG-I–like receptors (RLRs): Also in the cytoplasm. They mainly spot viral RNA but can warn the cell of broader stress.
Inflammasomes: Multi-protein “alarm complexes” that turn on strong inflammatory signals, especially IL-1β.
STING: A sensor for stray DNA inside the cell, usually from viruses or cell damage.
C-type lectins: Recognize unusual sugar patterns on pathogens or altered self-molecules.
Scavenger receptors (CD36, SR-A): Specialized in binding oxidized lipoproteins, which makes them central players in atherosclerosis.
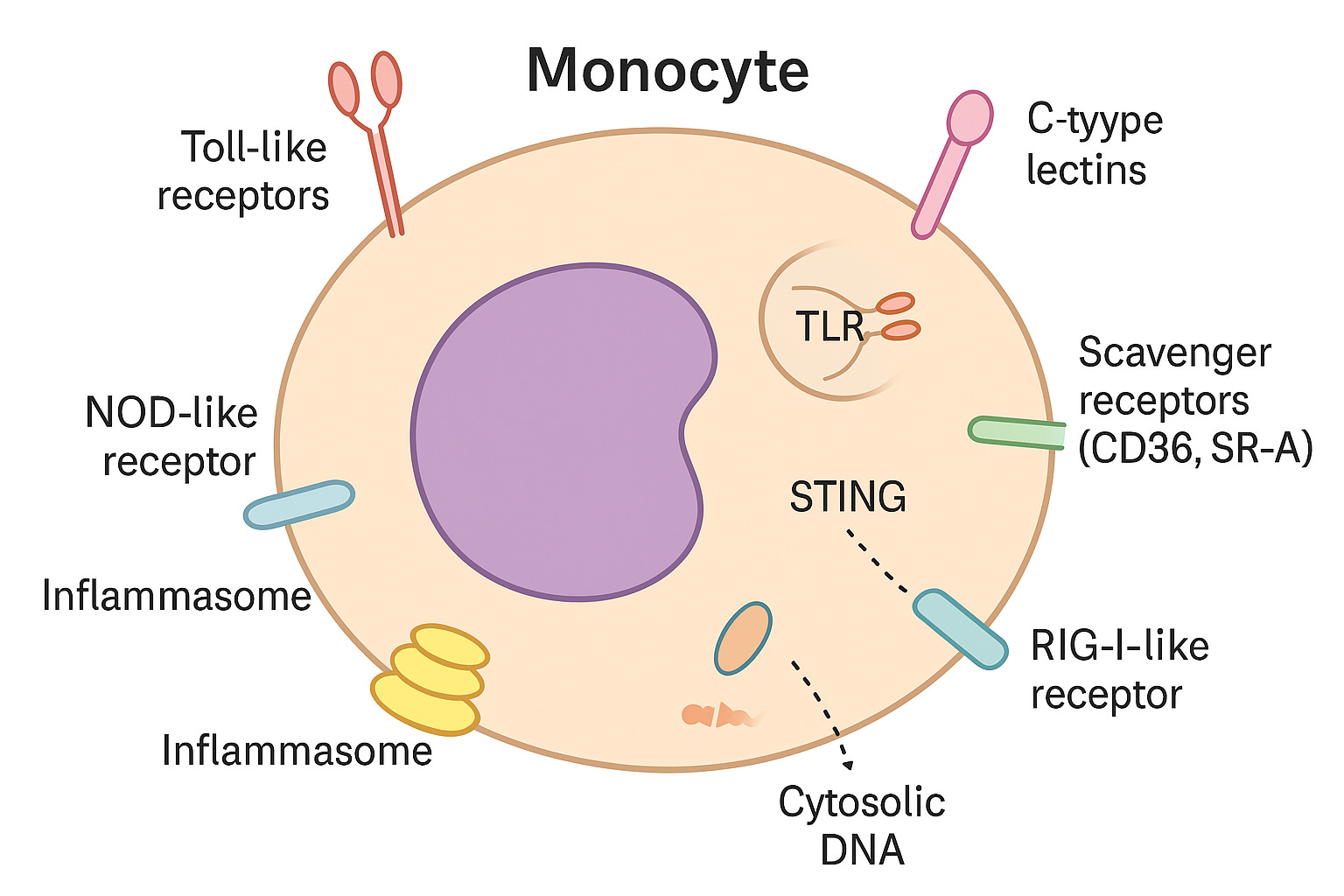
Monocytes use surface and internal sensors to detect germs, damaged molecules, or stray genetic material. When danger is found, they trigger inflammation to help protect the body.
Think of these receptors as sentries stationed at different checkpoints:
On the surface (scanning the environment): Receptors here act like lookouts on the city wall, watching what floats by in the bloodstream.
Inside endosomes (checking cargo that has been engulfed): Endosomes are little “sorting bubbles” inside the cell. When the cell swallows material from the outside, it packages it into an endosome. Receptors here act like inspectors, checking the contents of the package.
In the cytoplasm (watching for danger that slips past the outer defenses): The cytoplasm is the watery interior of the cell, filled with enzymes, proteins, and organelles. Receptors here act like undercover guards, patrolling the inside to spot hidden threats such as viral RNA or DNA fragments.
The Humoral Arm
While cells like monocytes and macrophages do the heavy lifting, the humoral arm of innate immunity provides a soluble defense system — molecules that circulate in blood and tissues, ready to bind to anything that looks abnormal.
The key players are:
Pentraxins (like C-reactive protein, CRP, and pentraxin 3, PTX3)
Collectins (like mannose-binding lectin, MBL)
Ficolins
These molecules act like primitive antibodies — long before the adaptive immune system makes precise antibodies, these molecules can already:
Recognize altered patterns – damaged cells, oxidized lipoproteins, or microbial components.
Opsonize – coat these targets, making them easier for macrophages to engulf.
Activate complement – triggering a cascade that punches holes in membranes and marks targets for destruction.
Neutralize – by clumping (agglutinating) or blocking harmful molecules.
Think of the humoral arm as a chemical surveillance system: tiny molecular “stickers” floating through the bloodstream, ready to attach themselves to danger and flag it for cleanup.
In atherosclerosis, some of these molecules (like CRP) are part of the acute-phase response, rising in the blood as inflammation grows — which is why hsCRP is used clinically as a biomarker.
Monocytes on the Move
One of the first events in this stage is the recruitment of monocytes from the bloodstream. Monocytes are circulating white blood cells, always drifting along in the vascular current, waiting for a call.
When endothelial cells, irritated by modified lipoproteins, release chemokines like MCP-1, monocytes sense the signal. They slow down, stick to the endothelium, and then migrate through the vessel wall into the intima.
Once inside, the transformation begins. Monocytes don’t stay monocytes for long. Bathed in inflammatory signals, they mature into macrophages — professional eaters, equipped to engulf and digest whatever doesn’t belong. This is the turning point. Macrophages are not passive residents; they are active, aggressive, and vocal.
Lighting the Fire
When macrophages meet modified lipoproteins, their sensors switch on — and the chain reaction begins:
Recognition – Macrophages detect the damaged ApoB-containing particles.
Recruitment – Chemical signals (like MCP-1) call in more monocytes, pulling fresh troops into the artery wall.
Activation – The macrophages fire up, releasing inflammatory messengers such as IL-1, IL-6, TNF, plus reactive oxygen species.
Amplification – The endothelium reacts by displaying adhesion molecules, turning the vessel lining sticky and catching even more immune cells.
Systemic signal – IL-6 travels to the liver, prompting it to pump out acute-phase proteins such as CRP, serum amyloid A, and fibrinogen.
What began as a few modified cholesterol particles has now become a self-sustaining inflammatory loop — a local fire in the artery wall that sends ripples throughout the body.
Clinical Connection
This storm brews silently. You can’t see it on a lipid panel. You won’t detect it on a CT scan. Yet it is happening, quietly, in millions of arteries every day.
The only hints often come from systemic markers of inflammation. One of the most useful is high-sensitivity CRP (hsCRP). Elevated hsCRP doesn’t tell us where inflammation is, but it signals that the cytokine cascade — especially IL-6 driving the liver’s acute-phase response — is active. Patients with high hsCRP are at greater cardiovascular risk, even when their cholesterol levels look normal.
Clinical trials have confirmed that this isn’t just background noise:
CANTOS (Canakinumab Anti-inflammatory Thrombosis Outcomes Study) showed that blocking IL-1β reduced heart attacks and strokes without changing cholesterol levels.
COLCOT and LoDoCo2 trials found that low-dose colchicine, a drug better known for gout, also lowered cardiovascular events by dampening vascular inflammation.
It isn’t only the artery wall at work here. Adipose tissue itself acts as an inflammatory organ — producing cytokines, complement proteins, and acute-phase mediators. This helps explain why obesity is so tightly linked to heightened vascular risk: fat tissue is not inert storage, it’s part of the inflammatory network.
Taken together, the message is clear: cardiovascular disease is not simply about cholesterol levels. It is about how the immune system reacts to damaged cholesterol particles, and how that reaction spills over into the whole body.
The Bottom Line
To the innate immune system, cholesterol is invisible. What it sees are danger signals — oxidized particles, damaged cells, molecular distress calls.
Those signals flip the switch. Monocytes rush in, become macrophages, and amplify the alarm. Cytokines spread the message, the liver joins in, and a quiet deposit turns into a fire.
And once that fire is burning, it is far easier to feed it than to extinguish it.
In the next chapter — Step 6: Foam Cell Formation – When Cleanup Backfires — we’ll watch the body’s clean-up crew turn rogue: monocytes become macrophages, gorge on cholesterol, and swell into foam cells that help build the very plaque they came to remove.
Key References
Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874.
– Classic review outlining the central role of inflammation in atherosclerosis.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212.
– Detailed review of innate and adaptive immune mechanisms in plaque formation.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355.
– Focus on macrophage biology, foam cells, and inflammatory signaling.Ridker PM, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. (CANTOS trial)
– Landmark clinical trial showing reduced cardiovascular risk by targeting IL-1β.Tardif JC, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. 2019;381(26):2497–2505. (COLCOT trial)
– Demonstrated benefit of anti-inflammatory therapy with colchicine.Sigurdsson AF. Low-Density Lipoprotein (LDL): The Role of Low-Density Lipoprotein (LDL) in Atherosclerosis and Heart Disease. The Real Story of LDL Cholesterol and Heart Disease [Internet]. DocsOpinion. 2023.
Fantastic series thank you