Step 6: Foam Cell Formation
When Cleanup Backfires

Atherosclerosis doesn’t appear overnight. It unfolds step by step, each injury deepening the one before it.
In Step 1, the endothelium faltered — the inner lining of the artery lost its protective barrier.
In Step 2, ApoB-containing lipoproteins slipped through that weakened barrier into the vessel wall.
In Step 3, those particles became trapped by sticky proteoglycans in the intima.
In Step 4, they underwent chemical modification, especially oxidation, which turned them toxic and recognizable as danger.
And in Step 5, the innate immune system answered the call, summoning monocytes into the arterial wall to help.
Now comes Step 6, where those very responders — macrophages — set out to clean the mess. But in a cruel twist, their attempt to heal the artery becomes the spark that drives the disease forward. This is the birth of the foam cell.
The Cleanup Crew
Monocytes migrate from the bloodstream into the intima, pulled by distress signals from the injured endothelium. Once inside, they transform into macrophages — the immune system’s garbage collectors.
In infection or wound healing, this role is heroic. Macrophages engulf microbes, digest debris, and restore order. But here, in the artery wall, they are confronted with something they are not built to handle: an endless tide of modified lipoproteins.
They mean well. They arrive as janitors. But their work will backfire.
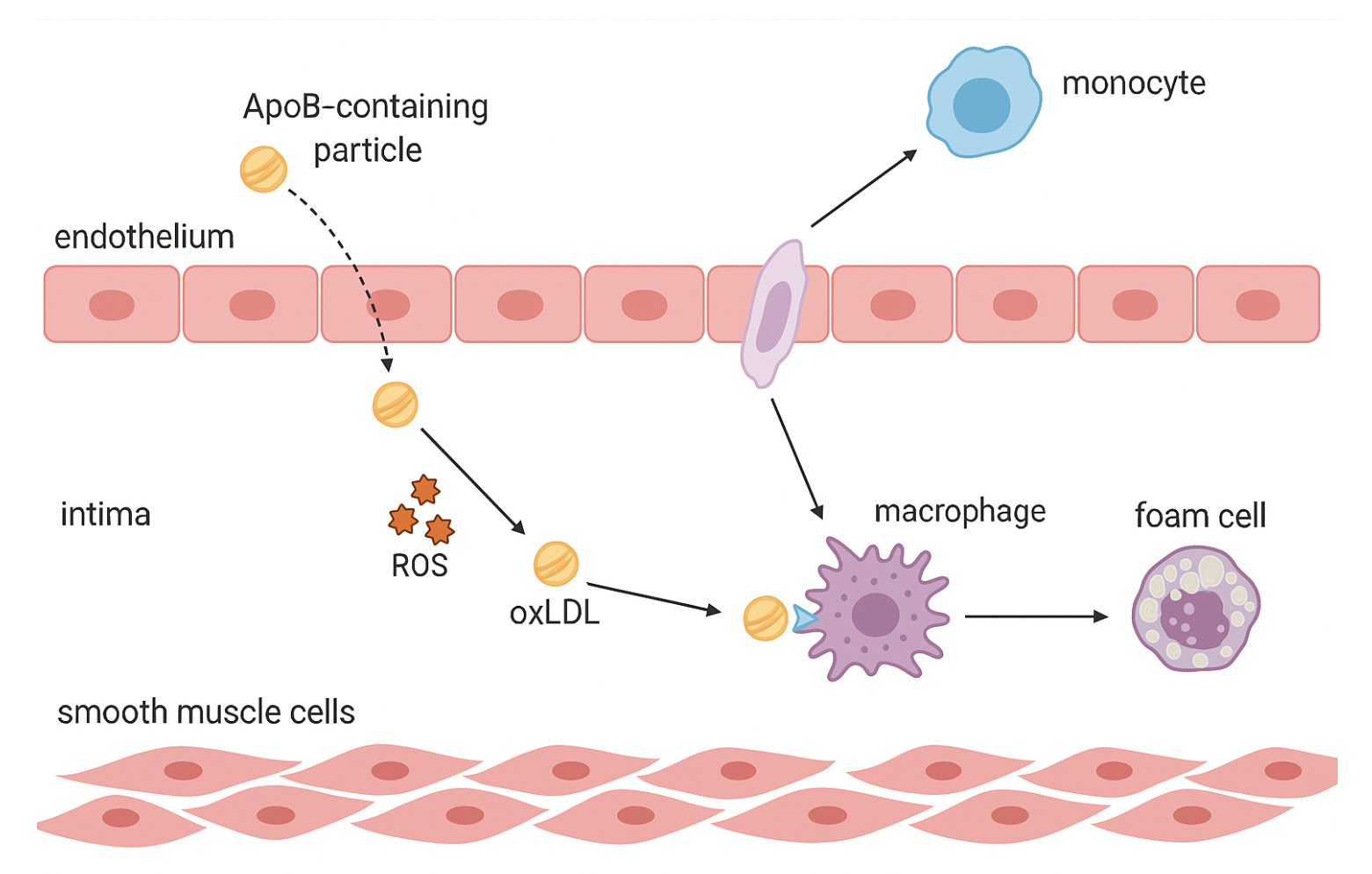
ApoB-containing particles cross the endothelium and become trapped in the intima. Once retained, they undergo oxidative modification. This triggers an inflammatory response, recruiting monocytes from the bloodstream. The monocytes migrate into the intima, transform into macrophages, and begin engulfing the modified particles, forming lipid-laden foam cells. This marks the early stage of atherosclerotic plaque development.
The Scavenger Trap
Macrophages take up oxidized lipoproteins through scavenger receptors like CD36, SR-A, and LOX-1. Unlike the LDL receptor, these have no “off switch.” There is no feedback inhibition to tell the cell it’s full.
The macrophages keep eating. Cholesterol droplets swell inside their cytoplasm, pushing the cell membrane outward until it resembles a bubble-filled sponge. Under the microscope, they look strangely beautiful, like cells packed with tiny pearls. Hence their name: foam cells.
What began as cleanup turns into overindulgence. And excess comes with consequences.
From Protectors to Provocateurs
Foam cells don’t just sit passively with their lipid cargo. They change character.
– They release cytokines — chemical messengers that summon even more immune cells into the lesion.
– They secrete enzymes that chew into the surrounding tissue, weakening the arterial wall.
– They generate reactive oxygen species, feeding the very oxidative stress that created the problem.
Instead of calming the fire, foam cells fan the flames. A defensive response becomes self-perpetuating damage.
Fatty Streaks: The Earliest Mark
As foam cells cluster, they create the first visible footprint of atherosclerosis: fatty streaks.
These yellowish streaks line the intima like paint strokes on the arterial wall. Autopsy studies have shown how early they appear. The Pathobiological Determinants of Atherosclerosis in Youth (PDAY) study found fatty streaks in teenagers, even in children. Nearly all young adults have them, particularly in the aorta and coronary arteries.
Not every fatty streak becomes a dangerous plaque. Some regress when risk factors are favorable. But they mark the beginning of something that, if unchecked, will claim more lives than any other disease.
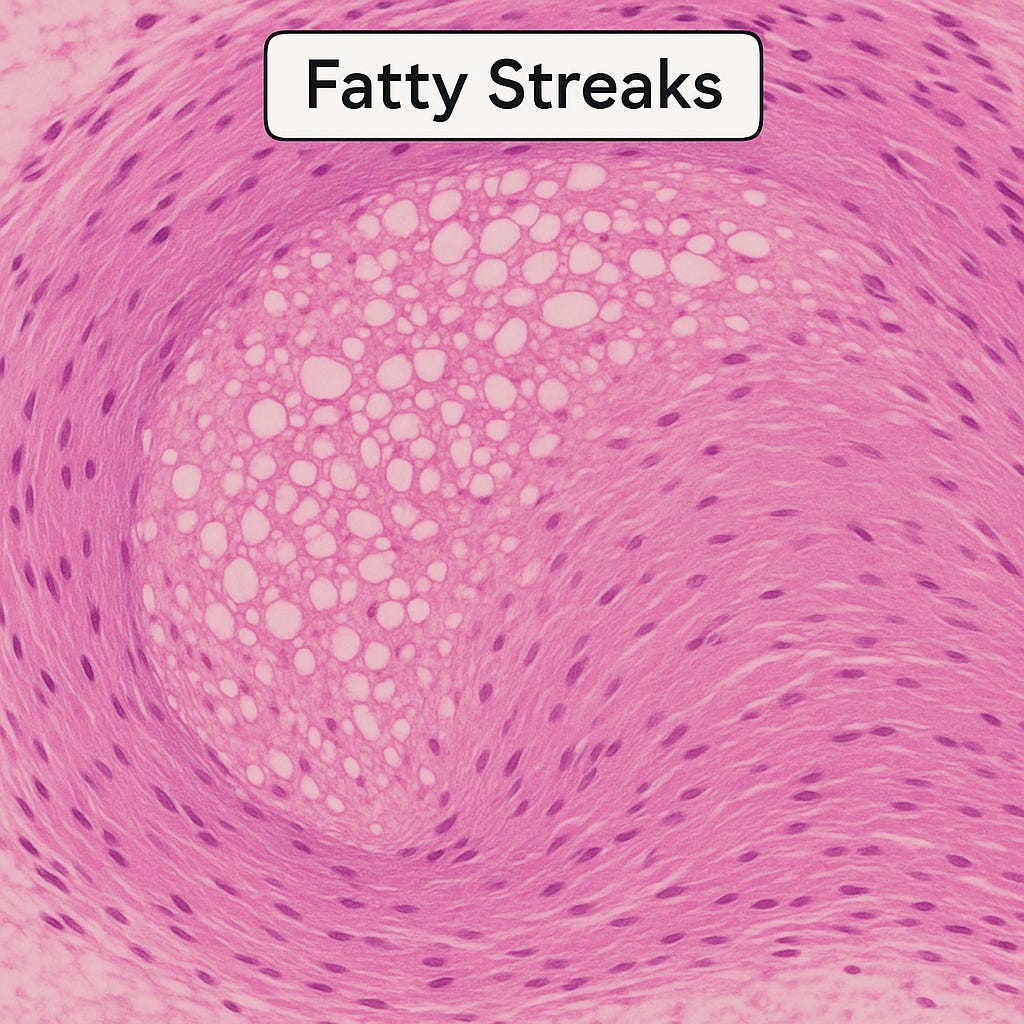
Histological view of a fatty streak: clusters of foam cells with vacuolated cytoplasm accumulate within the intima, forming the earliest visible lesion of atherosclerosis.
When Cleanup Fails
Foam cells cannot survive indefinitely. As they overfill, many undergo apoptosis — programmed cell death. Normally, dead cells are cleared quickly and quietly through efferocytosis, a housekeeping process where neighboring macrophages recycle the remains.
But in the hostile environment of an atherosclerotic plaque, efferocytosis falters. Dead foam cells are left to rot. Their contents spill into the intima: cholesterol crystals, oxidized lipids, ruptured membranes. This debris accumulates into the necrotic core, a toxic heart of the plaque.
The necrotic core does not sit harmlessly. It provokes more inflammation and destabilizes the thin fibrous cap above it. One day, that fragile barrier may rupture — exposing thrombogenic material to blood flow and triggering a heart attack or stroke.
What began as a cleanup may become a catastrophe.
Why Foam Cells Matter
Foam cells are invisible to scans. CT calcium scores, coronary angiography, even high-resolution imaging cannot show them. Yet their presence is nearly universal wherever atherosclerosis begins.
They remind us of two central truths:
– Particle burden drives the disease. The more ApoB-containing lipoproteins cross the endothelium, the more foam cells appear.
– Modification matters. It is not native LDL that macrophages devour, but oxidized, damaged forms — lipoproteins that the body mistakes for invaders.
This is why simply lowering LDL-C, while useful, tells only part of the story. What truly matters is reducing the number of circulating ApoB particles and minimizing the oxidative stress that distorts them.
Foam cells also point toward the future. Therapies that stabilize macrophages, reduce their inflammatory output, or improve efferocytosis may one day prevent the transition from fatty streak to necrotic core.
Bottom Line
Foam cells are the pivot point of atherosclerosis. They begin as janitors, arrive with good intentions, and end as saboteurs. With every engulfed particle, they dig the hole deeper.
Their formation marks the moment when atherosclerosis stops being a passive accumulation of lipoprotein particles and cholesterol and becomes an active, self-propelling disease.
Foam cells are where cleanup backfires — and where the silent seed of plaque begins to take root.
Next - Step-6 : Smooth Muscle Cell Migration and Cap Formation - Building the Wall
References
Ross R. Atherosclerosis — an inflammatory disease. N Engl J Med. 1999;340:115–126.
Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56.
Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118(4):653–667.
Stary HC. Natural history and histological classification of atherosclerotic lesions: an update. Arterioscler Thromb Vasc Biol. 2000;20:1177–1178.
McMahan CA, Gidding SS, Malcom GT, Tracy RE, Strong JP, McGill HC Jr; Pathobiological Determinants of Atherosclerosis in Youth Research Group. Pathobiological determinants of atherosclerosis in youth risk scores are associated with early and advanced atherosclerosis. Pediatrics. 2006 Oct;118(4):1447-55.
Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264.